RITE Exam
RITE (Residency In Training Exam)
What is RITE Exam:
- RITE is an exam held for all neurology residents PGY2-PGY4 in Feburary of every year.
Structure:
-
It consists of 425-Question test over 3.5 hours, divided as follows:
- Anatomy 10%
- Behavioral/Psych 10%
- Clinical adult 20%
- Clinical pediatrics 10%
- Contemporary issues 3%
- Neuroimaging 13%
- Pathology 10%
- Physiology 14%
-
Distributed over 3 sections:
- 1st and 3rd – Multiple choice
- 2nd – Imaging section
- Pathology
- CT, MRI
- EEG
- EMG
Aim:
- It is aimed to show you your areas of strength and weakness in order to prepare you for the board exam.
- For the program directors, it shows them if the program is successful in enriching the residents with the skills they need.
Importance for the Neurology board:
- The RITE scores correlate with the ABPN pass rate, as follows:
- 100% of those who scored in RITE > 75% (correct answers), passed the ABPN board on first attempt.
- 99% of those who scored in RITE > 65% (correct answers), passed the ABPN board on first attempt.
- Bottom line: if you score > 65% correct answers, you have high chances to pass the ABPN board exam.
Is the RITE exam similar to ABPN exam?
- NOT AT ALL
- RITE is more directed towards basic science, neurology board is directed to clinical science
- Very little of RITE questions will be encountered in board exam
- Neurology boards are WAY easier than RITE
- ABPN pass rate in 2020 was 91%
RITE Score Report:
- You will get report as "Percentile Rank" and "Percent Correct Answers"
- If you are in the 99 percentile per your class, this means you are one of the highest scoring 10 residents per your class (throughout US, 1000 neurology resident per PG year).

tPA for patients on DOAC
I went through this before when I had stroke patient with altered mentation, we were not able to tell if he was taking his Eliquis or not. So here is what I’ve gathered from that time.
There are two situations for patient on DOACs coming with acute stroke:
1- If they are not able to tell if they take their medications, as in dementia or obtunded patients. We can test for Anti Factor Xa, if normal (0), it means they haven’t taken their DOACs.
2019 guidelines states:“Alteplase could be considered when appropriate laboratory tests such as aPTT, INR, ecarin clotting time, thrombin time, or direct factor Xa activity assays are normal or when the patient has not taken a dose of these ACs for >48 h and renal function is normal.” –> So, factor Xa assay can be used to decide if they can get tPA.
2- if we know patient has been taking it and coming with severely disabling stroke. We can test factor Xa to tell if the medication is still working and their blood is still ‘thin’ (which precludes tPA) or not. This may be more relevant if the last dose was > 12h ago (half life for DOACs is around 12h). Since it is not a standard of care yet, will have to discuss it with family.
There are two types of Factor Xa assay, the general (heparin-calibrated) which is available in all hospitals and results are expressed in IU/ml, usually can be done within 10-15 minutes and the apixaban-calibrated or rivaroxaban-calibrated factor Xa with results expressed in ng/ml. Different studies showed that there is variation in DOACs trough levels. Here for example, rivaroxaban trough ranged from9.02‐147 ng/m. Levels < 30ng/ml is considered not clinically significant by many trials. Some trials as Annexa4, they didn’t give reversal agent Andexxa to patients with Anti-Xa levels <75 ng/ml presenting with major bleeding.
If DOAC-calibrated anti-Xa tests are not available, can we use the heparin-calibrated anti-Xa tests instead?
There has been multiple studies that compared the general Xa-assay (heparin calibrated) to DOAC-calibrated Xa and showed positive correlation.https://pubmed.ncbi.nlm.nih.gov/27377884/
https://pubmed.ncbi.nlm.nih.gov/32833716/https://pubmed.ncbi.nlm.nih.gov/30371316/-They compared heparin and DOAC-calibrated Xa, using cut-off of <0.3 IU/ml ruled out relevant DOAC concentrations with sensitivity of 92%. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6923159/– Japanese guidelines implementing the same principle of testing for coagulation profile (table 4) and giving tPA accordingly.
So, bottom line, if a patient on DOAC presented with severe stroke, last dose of DOAC was >12h then we may test Xa assay (available within 15 minutes). Levels < 0.3 IU/ml may indicate absence of clinically significant concentration.
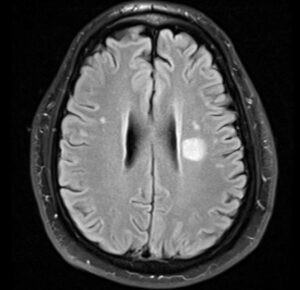
The Great Mimicker
The ever lasting question, Is it lymphoma?

A Puzzling case!
Be prepared, this patient I don't have a diagnosis yet!
She is a 60-year-old with known past medical history of HLD and CKD. At her baseline before all symptoms started she had a normal cognitive function, no weakness no seizures, basically she was normal.
Symptoms started in October with nausea & vomiting for which she was admitted to a local hospital with negative workup. While there she developed DVT and was started on Eliquis. She was admitted walking and was discharged on a wheel chair. Family reported she started to have generalized weakness, mainly in her legs when these symptoms started. Couple weeks later with progressive weakness she presented to our ED.
Here, She was found to have areflexive weakness in both upper and lower extremity more in lower extremities with 2/5 muscle power. LP did show markedly elevated proteins (103 mg/dl) with normal cell count so a diagnosis of GBS was entertained and the patient was started on PLEX with minimal improvement.
At the end of PLEX course patient started to show gradual progressive cognitive decline and disorientation. MRI brain with contrast was normal. This was blamed on UTI that was treated with antibiotics and patient was discharged to inpatient rehab on 10/22.
While in rehab, patient continued to progress with increasing confusion, disorientation and abnormal movements (choreo-athetosis) involving both upper extremities. Neurology was called and asked to transfer patient back for further workup for encephalopathy. A repeat MRI brain with contrast was still unremarkable. LP was again negative for infections with normal cell count and elevated protein. IgG synthesis was normal but had only 2 oligoclonal bands unique to CSF.
Is it an encephalopathy related to Guillain Bare as in Bickerstaff encephalitis, or it is really not Guillain Bare and it may be all paraneoplastic neuropathy and encephalopathy or autoimmune disease with these elevated protein and 2 oligoclonal bands? She had significant GI symptoms prior to all of her symptoms, can this be part of DPPH VGKC?
Further tests were sent for Anti Gq1b, anti gangliosides, 14-3-3 and autoimmune encephalopathy panel (Mayo clinic ENC1). CT chest, abdomen and pelvis were negative. Patient was started on pulse steroids for empirical treatment of autoimmune encephalopathy without any improvement.
Exam:
- Awake, alert, not attentive, not oriented to time, place or situation.
- Speech markedly slurred, incomprehensible in most part. Still able to follow simple commands though.
- No cranial nerve involvement. Normal EOM with no pursuit movement defect.
- Marked weakness 3/5 in both upper extremities and 0/5 in lower extremities with diffuse hypotonia and areflexia. Mute planter response.
- Abnormal movements, choreoathetotic movements in both upper extremities.
- Negative hyperekeplexia
Workup so far:
- MRI brain with contrast on 10/08 - 10/14 and 11/01 unremarkable. No restricted diffusion, no cortical ribboning and no white matter signal abnormality, no significant brain atrophy.
- CT chest, abdomen, pelvis unremarkable
- CSF: 10/09 with 1 WBC - 2 RBC - 60 glucose - 103 protein. 11/02 with 1 WBC - 44 RBC - 58 glucose - 125 protein - 2 OCB (7 CSF and 9 serum) - normal IgG index and synthesis rate - negative meningitis PCR panel
- TSH, ammonia, B12, B1 and electrolytes were all normal
- SPEP with IFA: unremarkable
- B1 53 (mildly decreased -> repleted) - Copper 1.59 (mildly elevated) -
- HIV, HTLV, crypto Ag and PCR negative
More advanced tests:
- Ganglioside panel negative for GQ1b but positive for GD1b (GD1b is associated with Guillain Bare, GQ1b is associated with Bickerstaff encephalitis)
- Autoimmune encephalopathy panel in CSF and serum negative
- Paraneoplastic panel negative
- Anti MOG negative
- Anti MAG negative
- CSF tau elevated, 14-3-3 came back elevated but CSF RT-QuIC negative
- EEG x2 with one prolonged: negative apart from delta-theta slowing - no PSWC
My working differentials were:
- Guillan Bare that developed Bickerstaff encephalitis: no pyramidal signs, no cranial nerve involvement, negative GQ1b.
- Autoimmune encephalopathy: Still possible although ENC1 and ENS1 panels were negative. No response to steroids or PLEX. Normal MRI brain, no seizures.
- Paraneoplastic encephalopathy: Still possible although CT chest/abdomen/pelvis are negative. Paraneoplastic and autoimmune encephalopathy panel in serum and CSF (which includes paraneoplastic causes as well) negative
- CJD: that was the diagnosis I was waiting for. MRI and EEG findings are usually negative in the beginning of the disease so I ordered 14-3-3 which came back positive with elevated Tau, however RT-QuIC is negative !!
What do you think?
Any other differentials I should have looked at?
What should we make with the 14-3-3 result with negative Rt-QuIC, may be she needs olfactory mucosa RT-QuIC? but how it is going to change treatment?

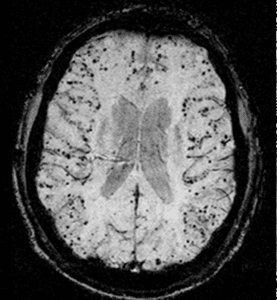
Amyloid Spells – A brain mine
![By Marvin 101 [CC BY 3.0 (https://creativecommons.org/licenses/by/3.0)], from Wikimedia Commons](https://neurologyresidents.com/wp-content/uploads/bb-plugin/cache/Cerebral_amyloid_angiopathy_CAA-MRI-landscape-feda5685e321ed812b6f6b2e76ed4b88-5b82dfebd5f4b.png "Cerebral_amyloid_angiopathy_(CAA)-MRI")
Clinical scenario:
Have you ever encountered an elderly patient with transient tingling, numbness or mild weakness and CT of the head or MRI of the brain showing a small cortical subarachnoid hemorrhage?
This used to be a dilemma, should we start the whole workup for SAH including CT angiography, conventional angiography and starting patient of nimodipine? The decision gets more difficult when you examine the patient, normal neurological exam or minor sensory deficit but you are stuck with this CT or MRI reporting subarachnoid hemorrhage.
When I had my first case, I felt we shouldn't expose the patient to invasive workup, serious medications and expensive stay at the stroke unit for a dot of cortical blood. The location of the blood doesn't fit with aneurysmal leakage and the patient reports no history of head trauma. When I encountered my second patient which was, surprisingly enough, in the same week, I felt there is something I'm missing or I need to read about. I looked it up and learned about this interesting presentation. After reading many papers, the conclusion was no workup is needed and no preventive measures are available. At that time, I was confident to discharge the patient from ED after having to explain to the emergency physicians, it is blood in subarachnoid space but it is not SAH!
Now, there is more studies talking about this clinical scenario with pathological reports demonstrating amyloid angiopathy as prevailing etiology.
Before talking about amyloid spells, let's first explain how cerebral amyloid angiopathy occurs:
- Cerebral amyloid angiopathy (CAA) is different from amyloidosis and not necessarily associated with Alzheimer disease as some people may think. These three are separate disorders with some similarities.
- Cerebral amyloid angiopathy as the name implies, is due to deposition of beta amyloid in the walls of leptomeningeal and cortical blood vessels. Most patients with CAA are sporadic, mainly due to impaired clearance of beta amyloid. Why the clearance is impaired, we don't have a satisfying answer yet. The other less common form is familial CAA in which different genes are implicated. Some mutations may give rise to both familial Alzheimer disease (AD) and CAA, which is seen in the Flemish, Dutch, Italian and Arctic CAA due to amyloid precursor protein (APP) mutation.
- CAA tends to involve more cortical and leptomeningeal blood vessels which explains the cortical location of both ICH and SAH seen in patients with CAA. Knowing that amyloid is cleared through the interstitial fluid tracks along the perivascular spaces, it explains why the most distal arteries (cortical and leptomeningeal) are more involved (1).
- Most patients with AD will have CAA but only 50% of CAA will have dementia. Which means AD involves beta amyloid deposition in both interstitial spaces and walls of blood vessels, so most AD will have CAA. CAA on the other hand can be isolated without interstitial beta amyloid deposition and without dementia or mild cognitive impairment.
Amyloid Spells:
- A transient positive or negative focal neurological symptom, in elderly patients (usually > 60-year-old) that correlates with cortical SAH or superficial cortical siderosis seen on imaging. Patients may report transient tingling that tends to migrate (positive symptom due to cortical irritation) or numbness and or weakness. Imaging reveals small amount of blood in the cortical subarachnoid space with MRI may show an evidence of cortical micro bleeds or superficial siderosis correlating with CAA (2).
Although there is no treatment for amyloid spells and no preventive measures available against further spells, it is usually associated with a higher risk for developing lobar ICH and future disability. A study (3) in 2015 involved 22 patients with cortical atraumatic SAH found that all of them fulfilled Boston criteria for CAA. Over a period of 30 months, out of the 22 patients, 5 patients have died. Out of the 17 survivors, 6 patients developed ICH, 12 patients developed cognitive impairment and 6 patients developed disability which shows us
Key Points:
- CAA is a common cause of transient focal neurological symptom in elderly patients, other causes may include TIA, migraine or seizures.
- You should consider CAA as the possible etiology if imaging revealed a small cortical SAH or evidence of cortical micro-bleeds on MRI.
- Keep other causes of cortical SAH in mind, including head trauma, cortical vein thrombosis.
- Identifying amyloid spell may protect the patient from un-needed, usually invasive workup and saves the patient's money.
- CAA is a brain mine, associated with increased risk of ICH and future disability, unfortunately no preventive measures are available yet.
References:
1- Weller RO, Massey A, Newman TA, Hutchings M, Kuo Y-M, Roher AE. Cerebral Amyloid Angiopathy : Amyloid β Accumulates in Putative Interstitial Fluid Drainage Pathways in Alzheimer’s Disease. The American Journal of Pathology. 1998;153(3):725-733.
2- Kumar S, Goddeau RP Jr, Selim MH, Thomas A, Schlaug G, Alhazzani A, Searls DE, Caplan LR. Atraumatic convexal subarachnoid hemorrhage: clinical presentation, imaging patterns, and etiologies. Neurology. 2010 Mar 16; 74(11):893-9.

Neurology Lectures
Neurological Examination Channels:


Univ. of Nebraska Neurological Examination - Adult
One of the best websites/channels, detailing all you need to know about neurological examination by Dr. Paul Larson.

Univ. of Nebraska Neurological Examination - Pediatric
One of the best websites/channels, detailing all you need to know about neurological examination by Dr. Paul Larson.
NeuroRadiology


Neuroradiology Advanced - LNR Course
All what you need to know about neuroradiology
More directed to PGY3/4

Grand Rounds


UA Neurology Grand Rounds
University of Arizona, Neurology Department grand rounds iTunes U channel.
You will need to download iTunes U then search for Neurology grand rounds

Audio/Video Lectures

Neurology - Podcast
Weekly podcast of content from Neurology®, the official journal of the American Academy of Neurology.
Continuum Audio Digest
If you don't have the time to read, then you can listen. Listen to lectures highlighting the important parts in the bi-monthly Continuum.
Free access for junior members, follow the link for more details.
Clinical Neurology Courses
Coursera - Medical Neuroscience, Duke University
An introduction of Neuroscience from Duke university. Starts with neuroanatomy and takes you through the physiology and how the brain works.

MDS Fundamentals Course
Movement disorders fundamental course by MDS (Movement Disorders Society)
Membership of MDS is free for neurology residents. Click here to proceed with free subscription.
Harvard University Fundamentals of Neuroscience
Fundamentals of Neuroscience, a free online course sponsored by Harvard School of Medicine
EEG Courses


USF EEG Fellowship Course
Online EEG course by USF and EEGCare. Usually held July-December.
About 3000 for physicians - 1500 for residents
NeuroIimmunology Resources


USF EEG Fellowship Course
Online EEG course by USF and EEGCare. Usually held July-December.
About 3000 for physicians - 1500 for residents
Video Galleries




Comments Section
Time for Feedback, Comments & Suggestions:
It has been about 6 months since the website was launched, at this point your feedback is vital for guidance towards further improvements.
Please, provide us with your comments, what did you like, what you didn't like or any suggestions in the comment section below.......
